


[주간 유전자] — 희귀병 분야 INPP5E 타겟팅






유럽연합은 희귀질환을 일반 인구 2,000명 중 1명 미만에게 영향을 미치는 질환으로 정의합니다. 현재 알려진 희귀질환은 6,000종 이상이며, 의학 문헌을 통해 새로운 질환이 지속적으로 보고되고 있습니다. 하지만 이러한 희귀질환은 인구의 상당 부분에 영향을 미치며, 대략 17명 중 1명이 평생 한 번 이상 희귀질환을 앓게 됩니다. 대부분의 희귀질환은 현재 효과적인 치료법이 존재하지 않습니다 [1].
사이아젠은 희귀질환 연구를 지원하며 다양한 마우스 및 랫트 모델 개발 서비스를 제공합니다. 이 글에서는 INPP5E 유전자와 관련된 희귀질환에 초점을 맞춰 설명하며, 관련 연구자들이 기존의 과학 문헌과 지식을 바탕으로 연구를 계속할 수 있도록 돕는 것을 목표로 합니다.
INPP5E 유전자 개요
INPP5E(Inositol Polyphosphate-5-Phosphatase E)는 단백질을 코딩하는 유전자입니다. INPP5E와 관련된 질환으로는 주버트 증후군 1형과 정신지체, 몸통 비만, 망막 이영양증, 소음경 증후군(MORM 증후군)이 있습니다.
이 유전자가 코딩하는 단백질은 이노시톨 1,4,5-트리스포스페이트(InsP3) 5-포스파타아제입니다. InsP3 5-포스파타아제는 세포 내 칼슘을 동원하고 다양한 자극에 대한 세포 반응을 매개하는 제2전달자인 Ins(1,4,5)P3를 가수분해합니다. 마우스 상동 유전자 연구에 따르면 이 단백질은 골지막 세포질 측에서 포스파티딜이노시톨 3,4,5-트리스포스페이트와 포스파티딜이노시톨 3,5-비스포스페이트를 가수분해하여 골지 소포 수송을 조절하는 것으로 추정됩니다. 이 유전자의 돌연변이는 중뇌-후뇌 기형과 망막 이영양증, 신장염증, 간 섬유화, 다지증 등을 포함한 다양한 섬모병증을 특징으로 하는 임상적·유전적으로 이질적인 질환군인 주버트 증후군을 유발합니다. 대체 스플라이싱은 서로 다른 이소폼을 코딩하는 여러 전사체 변이를 생성합니다 [2].
분자 기능 - 일차 섬모 조립에서의 Inpp5E
이전 연구들은 INPP5E가 섬모 기능 장애와 관련이 있음을 보여주었습니다 [3]. 일차 섬모(PC)는 세포와 생체 생리에 매우 중요한 원형질막 유래 구조입니다. 실제로 두 가지 5' PIP 포스파타아제인 Inpp5E와 Ocrl1의 결핍은 주버트/MORM 증후군과 같은 섬모병증 또는 로우 증후군과 같은 섬모병증 관련 질환과 명확히 연관되어 있습니다.
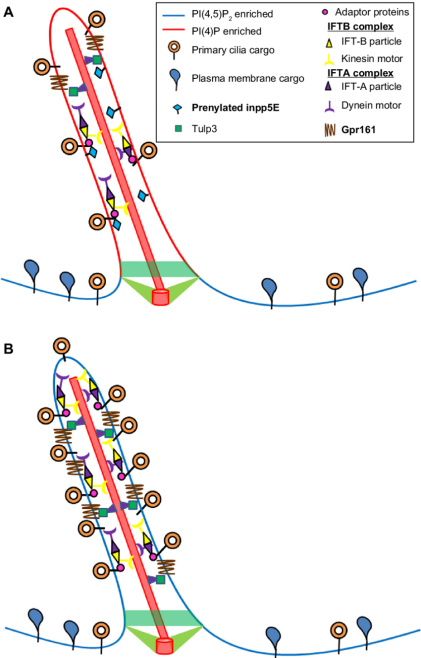
그림 1. Inpp5e의 섬모에서의 역할. 주석: (A) Inpp5e는 섬모 내에서 5' 포스파타아제로 기능하여 Pi(4,5)P2를 Pi(4)P로 전환합니다. 어댑터 단백질 Tulp3와 상호작용하여 섬모 화물을 수송하는 것으로 확인되었습니다. (B) 기능성 Inpp5e가 없는 경우 일차 섬모에 Pi(4,5)P2가 축적되고, 섬모가 섬모내 수송 입자와 어댑터 단백질을 격리합니다. 그 결과 섬모 분해가 영향을 받습니다 [3].
INPP5E 유전자 관련 희귀질환
주버트 증후군(JBTS)
주버트 증후군은 중뇌와 후뇌의 기형으로 인해 축방향 MRI에서 뇌간과 소뇌가 특징적인 어금니 모양으로 나타나는 신경발달 장애입니다. 핵심 임상 증상으로는 근긴장저하, 빈호흡/무호흡, 운동실조, 안구운동 실행증, 다양한 정도의 발달 지연 등이 있습니다. 또한 일부 환자는 망막 이영양증, 맥락망막 결손, 간신성 섬유낭포성 질환, 다지증 등을 동반합니다. 주버트 증후군은 유전적 이질성을 보이며, 일차 섬모의 발달과 안정성에 중요한 이노시톨 폴리포스페이트 5-포스파타아제 E를 코딩하는 INPP5E를 포함한 30개 이상의 유전자에서 돌연변이가 확인되었습니다 [4].
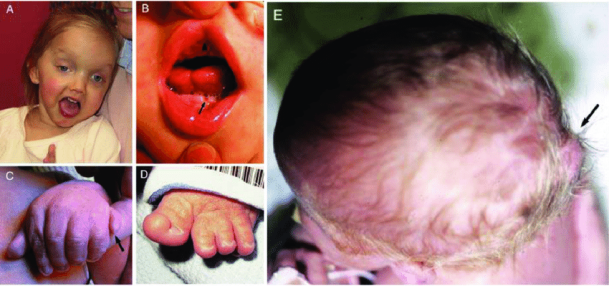
그림 2. 주버트 증후군의 임상 특징. (A) 27개월령 주버트 증후군(COACH 증후군) 여아의 얼굴 특징으로 넓은 이마, 아치형 눈썹, 사시, 오른쪽 눈 안검하수, 안면 긴장도 저하를 나타내는 벌어진 입 모습을 보입니다. (B) 주버트 증후군의 구강-안면-손가락 증후군 유사 특징을 보이는 소아의 구강 소견으로 정중선 상순 구순열(화살촉), 혀의 정중선 홈, 하악 치조 융기의 돌기(화살표)가 나타납니다. (C) 주버트 증후군과 축후 다지증을 가진 영아의 왼손(화살표). (D) 주버트 증후군과 엄지발가락 축전 다지증을 가진 영아의 왼발. (E) 두개골 후두부가 돌출된 작은 후두 뇌류를 가진 영아의 위에서 본 모습(화살표) [5].
간신성 섬유낭포성 질환(HRFCDs)
간신성 섬유낭포성 질환(HRFCDs)은 다낭성 신장과 선천성 간 섬유화와 같은 발달성 담문관 및 신장 섬유낭포성 이상을 특징으로 합니다. HRFCDs는 일차 섬모의 구조적 또는 기능적 결함으로 인해 발생하는 섬모병증이라는 더 큰 질환군에 속합니다. 섬모병증은 임상적·유전적으로 매우 이질적이며 선천성 발달 증후군으로 나타나거나 진행성 단일 장기 기능 부전으로 나타날 수 있는 지속적으로 확장되는 질환군입니다. 상염색체 열성 및 우성 다낭성 신장 질환(각각 ARPKD, ADPKD)은 인간에서 가장 흔한 HRFCD 형태입니다. 신장과 간 병변은 주버트 증후군, 메켈 증후군, 바르데-비들 증후군, 죤 증후군과 같은 다른 증후군성 섬모병증에서도 다양하게 발생합니다 [6]. Kati J. Dillard와 동료들은 INPP5E의 스플라이스 사이트 변이가 개에서 미만성 낭포성 신장 이형성증과 간 섬유화를 유발한다는 것을 발견했습니다.
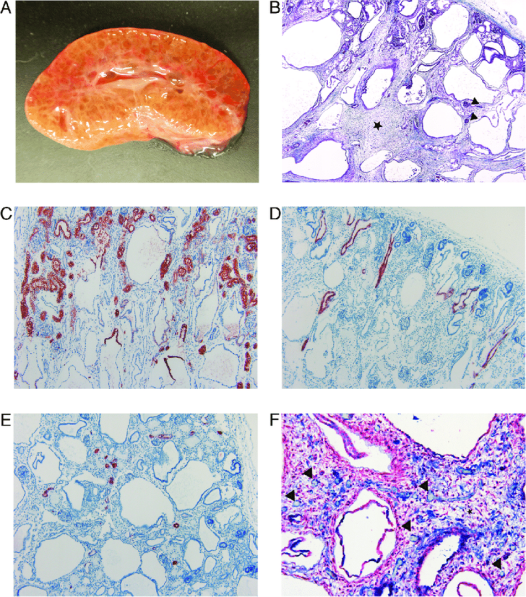
그림 3. 영향을 받은 신장의 거시적 및 조직학적 변화. (A) 영향을 받은 신장 단면 사진. 피질과 수질은 경계가 불분명하고 실질은 거의 전반적으로 낭포성입니다. (B) Masson 트리크롬 염색. 피질에 미성숙 사구체(검은색 화살촉)와 정상적인 세뇨관이 있습니다. 피질과 수질의 경계는 불분명합니다. 수질의 세뇨관은 확장되어 있으며 낭포성 세뇨관 사이에 느슨한 결합 조직(검은색 별표)이 증가되어 있습니다(4X). (C) 근위 세뇨관의 구불구불한 부분과 직선 부분 그리고 헨레 고리의 얇은 하행지의 상피 세포 마커인 AQP-1에 대한 IHC 염색(빨간색). 근위 세뇨관의 구불구불한 부분은 형태학적으로 정상으로 보입니다. 낭종의 상부에서 상피가 양성으로 염색되어 근위 세뇨관의 직선 부분과 헨레 고리의 얇은 하행지에서 기원했음을 나타냅니다. 낭종의 하부는 염색되지 않았습니다(10X). (D) 집합관 상피 세포 마커인 AQP-2에 대한 IHC 염색(빨간색). 집합관은 대부분 정상입니다. 피질에 약간 확장된 집합관이 몇 개 있습니다(10X). (E) 원위 구불세뇨관 상피 세포 마커인 칼빈딘-D28K에 대한 IHC 염색(빨간색). 원위 구불세뇨관은 형태학적으로 정상입니다(10X). (F) 중간엽 세포에 대한 α-SMA(빨간색)와 내피 세포에 대한 vWF(파란색)의 IHC 이중 염색. 결합 조직(검은색 별표)에 산재한 모세혈관(검은색 화살촉)이 포함되어 있습니다. 세뇨관과 가까이 있는 모세혈관은 매우 적습니다(20X) [7].
이 연구는 INPP5E 변이를 가진 인간과 노리치 테리어 간의 표현형 차이를 탐구합니다. 이 연구에서 확인된 변이는 인간 JBTS1 환자와 유사하게 개 INPP5E의 IPPc 도메인에 위치하지만, 스플라이스 사이트 변이는 현재까지 인간에서는 보고되지 않았습니다. 향후 종 간 스플라이스 사이트 변이와 관련된 추가적인 발견이 있을 수 있습니다.
원스톱 마우스 모델 검색 플랫폼: MouseAtlas
MouseAtlas는 KO부터 인간화 마우스까지 유전자 및 제품 모델명 검색만으로 원하는 모델을 쉽게 찾을 수 있는 플랫폼입니다. 생체 마우스인지 정자 상태인지, 실시간 재고 상황, 검증 데이터, 상세 설명 등을 직관적으로 확인하고 바로 주문할 수 있습니다. 당사 내부 제품 관리 시스템과 연동되어 실시간으로 업데이트되며, 현재 39,000종 이상의 모델 마우스가 등록되어 있어 연구자들에게 매우 편리한 원스톱 솔루션을 제공합니다.
계속해서 읽기: Inpp5e 마우스 모델
아래 다음 글에서는 Inpp5e 유전자 연구에 사용되는 마우스 모델의 응용 분야와 새로운 연구 성과에 대해 자세히 제공합니다.
>> 더 알아보기: Inpp5e 마우스 및 새로운 연구 성과
참고문헌
[1] European Commission-European Commission.(2020).Rare diseases.[online] Available at:https://ec.europa.eu/info/research-and-innovation/research-area/health/rare-diseases_en [Accessed 27 Jan.2020].
[2] https://www.genecards.org/cgi-bin/carddisp.pl?gene=INPP5E&keywords=INPP5E
[3] Madhivanan K,Ramadesika S, Aguilar R C. Role of Ocrl1 and Inpp5E in primary cilia assembly and maintenance: a phosphatidylinositol phosphatase relay system?[J]. Research and Reports in Biology, 2016, 7: 15-29.
[4] https://medlineplus.gov/genetics/condition/joubert-syndrome/
[5] Parisi M A.Clinical and molecular features of Joubert syndrome and related disorders[C]//American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Hoboken:Wiley Subscription Services, Inc.,A Wiley Company,2009,151(4): 326-340.
[6] Gunay‐Aygun, Meral. "Liver and kidney disease in ciliopathies." American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Vol.151.No. 4.Hoboken: Wiley Subscription Services,Inc., A Wiley Company, 2009.
[7] Dillard K J, Hytönen M K, Fischer D, et al. A splice site variant in INPP5E causes diffuse cystic renal dysplasia and hepatic fibrosis in dogs[J]. PloS one, 2018, 13(9): e0204073.



