


SYNGAP1 관련 지적장애 (MRD5)






유럽연합은 희귀질환을 일반 인구 2,000명 중 1명 미만에게 발병하는 질환으로 정의합니다. 현재 7,000여 종의 희귀질환이 존재하는 것으로 추정됩니다. 발병률이 낮음에도 불구하고 미국에서 희귀질환을 앓고 있는 환자의 총 수는 2,500만~3,000만 명으로 추산됩니다. 이러한 수치는 개별 질환은 드물 수 있지만 희귀질환 환자의 총 수는 많다는 것을 보여주며, 이는 희귀질환 커뮤니티에 익숙하지 않은 사람들이 종종 오해하는 사실입니다.
사이아젠은 희귀질환 연구를 지원하며 SYNGAP1 관련 설치류 동물 모델을 비롯한 다양한 마우스/랫트 모델을 제공합니다. 본 글은 SYNGAP1 유전자와 희귀질환 MRD5를 중심으로 설명하며, 관련 연구자들이 과학 문헌과 지식을 기반으로 연구를 계속할 수 있도록 돕는 것을 목표로 합니다.
SYNGAP1 유전자 개요
SYNGAP1(Synaptic Ras GTPase Activating Protein 1)은 단백질 코딩 유전자입니다. SYNGAP1 유전자는 6번 염색체에 위치하며 SynGAP 단백질을 생성하는 역할을 합니다. 이 단백질은 뉴런이 서로 통신하는 시냅스에서 조절자로 작용합니다. SYNGAP1 유전자의 변이로 인해 유전자가 충분한 SynGAP 단백질을 생성하지 못하게 됩니다. 적정량의 SynGAP 단백질이 부족하면 시냅스의 흥분성이 증가하여 뉴런이 효과적으로 통신하기 어려워지며, 이로 인해 SYNGAP1 돌연변이 환자에서 나타나는 많은 신경학적 문제가 발생합니다.
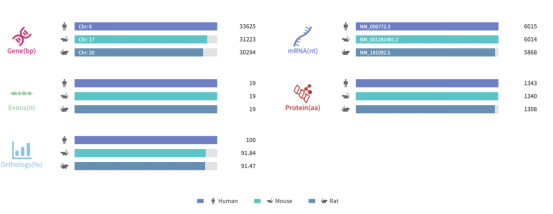
그림 1. SYNGAP1의 유전자 정보 및 서열 상동성 정렬
출처: Rare Diseases Data Center (RDDC)
SYNGAP1 유전자 관련 희귀질환
상염색체 우성 비증후군성 지적 장애 5 (MRD5)
MRD5(Mental Retardation, autosomal Dominant 5)는 희귀 비증후군성 지적 장애로 분류됩니다. MRD5는 주로 중추신경계에 영향을 미치는 SYNGAP1 유전자 돌연변이로 인해 발생하는 질환입니다. 주로 생후 첫 몇 년 동안 뚜렷하게 나타나는 중등도~중증의 지적 장애를 특징으로 합니다. 일부 환자는 발작 및/또는 자폐 스펙트럼 장애를 경험할 수도 있습니다. 보고된 거의 모든 사례는 새로운(de novo) 돌연변이로 인한 것이지만, 상염색체 우성 방식으로 후세대에 유전될 수도 있습니다. 치료는 각 환자에게 나타나는 징후와 증상에 따라 진행됩니다.[1][2][3]
MRD5의 분자 유전학
SYNGAP1 유전자의 반수체 부족(haploinsufficiency)을 유발하는 기능 소실 돌연변이(및 전뇌 글루타메이트성 뉴런의 발달을 손상시킬 가능성)가 이 질환의 원인입니다. 대부분의 돌연변이는 단백질 절단 돌연변이(무의미 돌연변이, 프레임시프트 돌연변이, 스플라이스 돌연변이)이며, 소수의 미스센스 돌연변이도 보고된 바 있습니다. 지금까지 보고된 돌연변이는 다음과 같습니다:
- c.412A>T (p.Lys138*),
- c.1735C>T (p.Arg579*) 및 c.2438delT (p.Leu813Argfs*23) [4],
- c.501-1G>A, c.2294+1G>A (13번 엑손의 스키핑 및 조기 종결 Glu706LeufsTer38 유발),
- c.1084T>C (p.Trp362Arg),
- c.1685C>T (p.Pro562Leu), [5]
- c.2212_2213del (p.Ser738*) (연구진에 따르면 질병 유발 가능성이 있음)[6],
- Trp267* 및 Arg143* [7],
- c.348C>A (p.Y116*) [8]
전체 유전자 결실 1건도 보고된 바 있습니다.[9] 실제로 MRD5는 6p21.3 미세결실(SYNGAP1을 포함한 여러 유전자의 결실 유발)과 임상적으로 중복될 수 있으며, 이는 유전자 손실도 반수체 부족의 원인이 될 수 있음을 확인시켜 줍니다. SYNGAP1과 여러 다른 유전자를 포함한 결실이 있는 환자도 자폐 스펙트럼 장애를 보일 수 있습니다.[10]
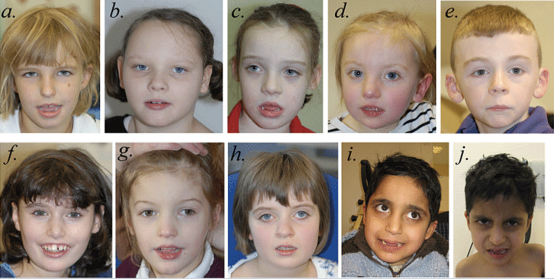
그림 2. SYNGAP1 반수체 부족 환자의 얼굴 특징. 7세 3개월 환자 1(a); 8세 2개월 환자 2(b); 7세 9개월 환자 3(c); 3세 2개월 환자 4(d); 8세 4개월 환자 5(e); 12세 10개월 환자 6(f); 5세 7개월 환자 7(g); 8세 7개월 환자 8(h); 8세 3개월 환자 9와 10(i 및 j)의 얼굴 사진. 가장 흔한 공통 얼굴 특징은 약간 아래로 기울어진 아몬드 모양의 눈꺼풀 틈입니다. 환자 5(e)를 제외한 나머지는 입을 벌리고 비교적 풍만한 아랫입술을 가진 경미한 근병증 모습을 보입니다. 환자 1(a), 3(c), 4(d), 6(f), 7(g), 9(i), 10(j)은 비교적 긴 코를 가지고 있으며, 환자 2(b), 4(d), 5(e), 6(f), 7(g), 8(h)은 돌출된 귓불이 있는 비교적 긴 귀를 가지고 있습니다. 환자 1(a), 6(f), 9(i), 10(j)은 비교적 깊게 들어간 눈을 가진 것으로 보이며, 환자 8(h)은 어느 정도 안검하수가 있습니다. 환자 6(f)은 외상으로 인해 중절치가缺失되어 있습니다. 이 시리즈에서 유일한 전체 유전자 결실 환자인 환자 7(g)가 다른 환자와 외모에서 크게 다르지 않다고 판단됩니다.[9]
연구 프로젝트 및 임상 시험
Orphanet 데이터베이스의 자료에 따르면 현재 MRD5에 대한 임상 시험은 없으며 26건의 연구 프로젝트가 진행 중입니다.

출처: Orphanet [11]
기타 관련 질환
- 근간대-무긴장 간질
- 비특이적 조기 발병 간질성 뇌병증
- SYNGAP1 관련 발달 및 간질성 뇌병증
SYNGAP1 유전자 돌연변이의 주요 증상
- 지적 장애 (경증~중증)
- 저긴장증 (근긴장 저하)
- 전반적 발달 지연
- 간질 (미세한 눈꺼풀 떨림, 간대성 근경련, 응시 발작 및 탈력 발작)
- 감각 처리 장애
- 대운동 및 미세 운동 기술 지연
- 운동실조 (협응 장애)
- 언어 지연/구어 실행증 (경증~중증)
- 자폐 스펙트럼 장애
- 수면 및 행동 장애
- 시각 이상
사이아젠의 지원 서비스
지난 1년간 사이아젠은 희귀질환 연구 커뮤니티를 지원하기 위한 교육 자원과 이니셔티브 개발에 집중해 왔습니다. 희귀질환 연구를 위한 정밀 동물 모델의 연구개발을 지원하기 위해 2021년 1분기에 희귀질환 모델 협력 프로그램을 시작했습니다. 이 프로그램을 통해 해당 분야의 발전에 필요한 차세대 동물 모델에 대한 아이디어를 가진 희귀질환 연구자 커뮤니티를 구축하는 것을 목표로 합니다.
원스톱 마우스 모델 검색 플랫폼: MouseAtlas
MouseAtlas는 KO부터 인간화 마우스까지 유전자 및 제품 모델명 검색만으로 원하는 모델을 쉽게 찾을 수 있는 플랫폼입니다. 생체 마우스인지 정자 상태인지, 실시간 재고 상황, 검증 데이터, 상세 설명 등을 직관적으로 확인하고 바로 주문할 수 있습니다. 당사 내부 제품 관리 시스템과 연동되어 실시간으로 업데이트되며, 현재 39,000종 이상의 모델 마우스가 등록되어 있어 연구자들에게 매우 편리한 원스톱 솔루션을 제공합니다.
참고문헌
1. Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot-Cerioli M, Lacaille JC, Niu Z, Eng CM, Yang Y, Palardy S, Belhumeur C, Rouleau GA, Tommerup N, Immken L, Beauchamp MH, Patel GS, Majewski J, Tarnopolsky MA, Scheffzek K, Hjalgrim H, Michaud JL, Di Cristo G. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat. 2013 Feb;34(2):385-94. doi: 10.1002/humu.22248. Epub 2012 Dec 12. PMID: 23161826.
2. Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, Joober R, Lacaille JC, Nadeau A, Milunsky JM, Wang Z, Carmant L, Mottron L, Beauchamp MH, Rouleau GA, Michaud JL. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry. 2011 May 1;69(9):898-901. doi: 10.1016/j.biopsych.2010.11.015. Epub 2011 Jan 15. PMID: 21237447.
3. Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Côté M, Perreau-Linck E, Carmant L, D'Anjou G, Fombonne E, Addington AM, Rapoport JL, Delisi LE, Krebs MO, Mouaffak F, Joober R, Mottron L, Drapeau P, Marineau C, Lafrenière RG, Lacaille JC, Rouleau GA, Michaud JL; Synapse to Disease Group. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009 Feb 5;360(6):599-605. doi: 10.1056/NEJMoa0805392. Erratum in: N Engl J Med. 2009 Oct 29;361(18):1814. Perreault-Linck, Elizabeth [corrected to Perreau-Linck, Elizabeth]. PMID: 19196676; PMCID: PMC2925262.
4. Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y, Higashi K, Park AR, Spiegelman D, Dobrzeniecka S, Piton A, Tomitori H, Daoud H, Massicotte C, Henrion E, Diallo O; S2D Group, Shekarabi M, Marineau C, Shevell M, Maranda B, Mitchell G, Nadeau A, D'Anjou G, Vanasse M, Srour M, Lafrenière RG, Drapeau P, Lacaille JC, Kim E, Lee JR, Igarashi K, Huganir RL, Rouleau GA, Michaud JL. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet. 2011 Mar 11;88(3):306-16. doi: 10.1016/j.ajhg.2011.02.001. Epub 2011 Mar 3. Erratum in: Am J Hum Genet. 2011 Apr 8;88(4):516. PMID: 21376300; PMCID: PMC3059427.
5. Berryer MH, Hamdan FF, Klitten LL, Møller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot-Cerioli M, Lacaille JC, Niu Z, Eng CM, Yang Y, Palardy S, Belhumeur C, Rouleau GA, Tommerup N, Immken L, Beauchamp MH, Patel GS, Majewski J, Tarnopolsky MA, Scheffzek K, Hjalgrim H, Michaud JL, Di Cristo G. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat. 2013 Feb;34(2):385-94. doi: 10.1002/humu.22248. Epub 2012 Dec 12. PMID: 23161826.
6. Volk A, Conboy E, Wical B, Patterson M, Kirmani S. Whole-Exome Sequencing in the Clinic: Lessons from Six Consecutive Cases from the Clinician's Perspective. Mol Syndromol. 2015 Feb;6(1):23-31. doi: 10.1159/000371598. Epub 2015 Feb 3. PMID: 25852444; PMCID: PMC4369115.
7. Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, Malone S, Wallace G, Stanley T, Bye AM, Bleasel A, Howell KB, Kivity S, Mackay MT, Rodriguez-Casero V, Webster R, Korczyn A, Afawi Z, Zelnick N, Lerman-Sagie T, Lev D, Møller RS, Gill D, Andrade DM, Freeman JL, Sadleir LG, Shendure J, Berkovic SF, Scheffer IE, Mefford HC. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013 Jul;45(7):825-30. doi: 10.1038/ng.2646. Epub 2013 May 26. PMID: 23708187; PMCID: PMC3704157.
8. von Stülpnagel C, Funke C, Haberl C, Hörtnagel K, Jüngling J, Weber YG, Staudt M, Kluger G. SYNGAP1 Mutation in Focal and Generalized Epilepsy: A Literature Overview and A Case Report with Special Aspects of the EEG. Neuropediatrics. 2015 Aug;46(4):287-91. doi: 10.1055/s-0035-1554098. Epub 2015 Jun 25. PMID: 26110312.
9. Parker MJ, Fryer AE, Shears DJ, Lachlan KL, McKee SA, Magee AC, Mohammed S, Vasudevan PC, Park SM, Benoit V, Lederer D, Maystadt I, Study D, FitzPatrick DR. De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am J Med Genet A. 2015 Oct;167A(10):2231-7. doi: 10.1002/ajmg.a.37189. Epub 2015 Jun 15. PMID: 26079862; PMCID: PMC4744742.
10. Cook EH Jr. De novo autosomal dominant mutation in SYNGAP1. Autism Res. 2011 Apr;4(2):155-6. doi: 10.1002/aur.198. PMID: 21480541.
11. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=101685



